2022年5月23日晚,君实生物发布公告称,VV116对比辉瑞Paxlovid早期治疗轻中度COVID-19的三期临床试验达到主要研究终点,将在近期递交上市申请。
按照君实生物相关人士对投资者的说法,这代表试验的成功。在业内人士看来,这一“成功”,很可能成为VV116通过国家药监局审批的助推剂——迄今为止,还没有一个国产新冠口服药通过药监局审批。而君实对比的辉瑞Paxlovid,是国家药监局唯一批准的小分子新冠口服药。
一些业内人士表示,这一临床设计有些“匪夷所思”,如果按照这一标准通过审批,那么意味着很多同类的药也可以按照此方案设计通过评审。受到VV116试验方案的“启发”,已经有一些后面还没有到三期临床的新冠口服药的公司,已经在找CRO设计同样的方案了。
在公告发布次日,君实生物股价暴跌,在科创板和港股价格跌幅一度分别超过15%和10%。
显然,君实生物公布的这一试验结果,并未说服投资者。
目前国际上市的新冠药物,如辉瑞Paxlovid、默沙东Molnupiravir,都是双盲对照试验。君实生物这一试验则是单盲。
此外,更重要的问题是:在昨夜的公告中,君实生物只表示达到了这一试验的主要终点“至持续临床恢复的时间”,而对次要终点并未提及。而这一“终点”,即重症和死亡率,在目前的国产新冠口服药的审评标准里更为重要。
但君实这一试验的次要研究终点,只是包括“截至第28天发生COVID-19进展(定义为进展为重度/危重COVID-19或全因死亡)的受试者百分比”等。
考虑到辉瑞的P药获批主要终点是围绕“重症改善率”和“预防死亡”的,P药并没有一些关于“轻中症改善”的数据,拿VV116和P药做针对“轻中症改善”的非劣实验,在一些人看来不够严谨。
但值得注意的是,此次君实在瑞金的头对头试验,并不是没经过CDE的讨论。
君实生物这次试验结果公布后,或许会有更多国内药企跟随其后,将临床恢复作为试验的主要终点。
不过,君实生物这一试验的未提及的次要终点,即重症和死亡率,在目前的审评标准里更为重要。
在奥密克戎传染性强、致病性弱的条件下,国内新冠口服药审批指导原则例强调的“重症改善率”,仍是阻碍很多新冠口服药的临床进度的一大难题。
01、“非劣性”试验方案设计颇受争议
在最新的极高感染率、极少重症患者的奥密克戎流行株面前,新冠口服药三期临床的国家药监局审批标准,依旧是以症状改善为“临床终点”,尤其是“降低低重/危重患者的死亡率”。
今年4月,君实生物在上海瑞金医院开展了一项和辉瑞新冠口服药paxlovid的头对头试验,4月底已完成患者入组。这项试验,据业内人士介绍,在相关部门的帮助下:进展极快,不管在审批速度还是不断更改的病人入组数量上,都有着超越一般流程的效率。
但这项“头对头”临床试验的方案设计,也颇受争议。
辉瑞的paxlovid,开展新冠口服药临床试验时,新冠疫苗在美国还没有大规模接种,当时的新冠病毒还没有产生像奥密克戎那样的变种,在美国特定的人群中导致的住院率和重症率都较高。辉瑞的临床测量目标包含住院患者在医院和重症监护室停留的天数、死亡患者的比例,这是paxlovid通过FDA审批的关键数据。
那么,在奥密克戎毒株感染的入组病人中,重症率极低且大多数可能接种了疫苗,那么这项“头对头试验”从入组人群来说已经和辉瑞的paxlovid的入组人群有了极大的不同。
新冠口服药三期临床的国家药监局审批标准,主要以症状改善为“临床终点”,但“降低低重/危重患者的死亡率”,对重症病人群体相对容易判断,而奥密克隆感染几乎没有重症患者,很难做出重症的统计学的差距。
一位业内人士表示,这项vv116和辉瑞p药的头对头试验,开始设计的临床终点是双终点:一个是患者转化成重症的重症率指标;第二个是高风险人群临床症状的改善。
但后来在试验过程中发现,不管是吃安慰剂、辉瑞的paxlovid还是VV116,重症率都是零——在几百个奥密克戎阳性感染病人中很难找到一个重症病人。
后来,双终点变成了“以症状改善为主的临床终点”,而且最重要的是,“以症状改善为主的临床终点”是一个和辉瑞paxlovid对比的“非劣性试验”,即不比辉瑞paxlovid差就可以。
但辉瑞paxlovid在“以症状改善为主要临床终点”的EPIC-SR的三期临床至今未取得成功,而paxlovid通过FDA审批通过的EPIC-HR的关键数据“包含住院患者在医院和重症监护室停留的天数、死亡患者的比例”。
“paxlovid没有做出症状改善和安慰剂的差异,按照一般的认为就是没有做出差距。paxlovid临床症状改善跟安慰剂组是完全一样13天,而且安慰剂比它好一点”。上述人士认为。
简而言之,paxlovid在症状改善上跟安慰剂是非劣,然后现在VV116跟paxlovid在症状改变上也去比非劣,最后的结果是什么?即VV116在“症状改善上的临床终点”和安慰剂比是“非劣”——即只是不比安慰剂差而已。
“这个设计是一个荒唐的设计,就是说假如这么能通过审批的话,后面的药任何其他的药都很容易获批。”
在奥密克戎毒株感染者中,非劣性试验很容易做。因为,奥密克戎感染者一般不用药,第5天就改善了,实际上跟安慰剂组实际上没有统计学差异。
那么,中国很多的那些抗核苷类的抗病毒的药都可以“以猫化虎”来设计。
新冠的症状改善是跟安慰剂的优效很难做,但要做非劣很容易。新冠的症状有十几个症状,有些症状是安慰剂组的效果会更好,比如说腹泻、肠胃道反应等。
02、疫情压力下,国产新冠口服药一路绿灯
公共卫生事件下的应急药品,核心要素就是快。
早在2021年上半年,国内医药行业一致预期是在中国的防疫政策下,不会出现大规模的新冠患者,因此也不需要大量的新冠治疗类药物;但随着病毒变种传染性加快,以及中国终归需要打开国门这两点,新冠治疗性药物也越来越被提上日程,尤其是给药方式更方便、可及性更高口服药。
君实是最早布局新冠口服药的企业之一。
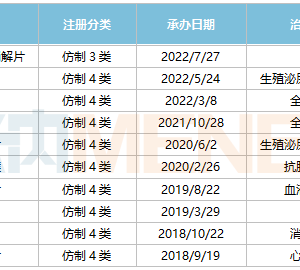
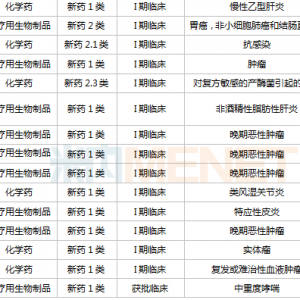
早在去年10月,君实官宣和旺山旺水达成全球除中亚五国外的合作,便启动了一系列临床,2021年年底在乌兹别克斯坦完成中重症的二期临床之后,很快收到了乌方的EUA,也成了国内首个在海外批准上市的新冠口服药。
今年开年以来,君实分别开展了一项重症标准治疗对照研究(也就是此次EMI发表的文章),以及其他三项轻中症状研究,这是除开拓和真实生物两款“成熟药改新冠适应症”之外进度最为靠前的本土新冠口服药了。
而在4月君实生物在瑞金开展的头对头试验里,据一份网传纪要透露,此次头对头试验临床观测终点有二点:特续临床恢复的时间,重症率转化和全因死亡(最新修改只保留了前者,即临床改善)。
试验最初设计样本是400多例,全部在瑞金医院,后来考虑到单中心数据说服力不够,又分别在新华等医院纳入不少新病例,已经有800多例入组——对人数样本的不断变化,一位业内人士认为,这在常规的临床实验中是不被允许的,但这次显然有“特事特办”的成分在里面。
因为被争议的“非劣”试验,是看统计学结果,加到一定的样本量,成功的概率会大。不断加样本量,是为了增加试验成功的概率。一位业内人士称。
此次试验虽然是单盲试验,但考虑到辉瑞的paxlovid已经在美国FDA和中国CDE都收到过EUA批件,并且成功进入医保,VV116的核心目标在于快速获批,而双盲设计需要更多的前期准备(比如把药片做得一模一样),单盲设计是在临床速度和研究质量上面更多偏向了前者。
据上述人士透露,此前,瑞金的这个头对头实验是一个IIT(研究者发起)试验,但后来改成了一个注册性临床,这么一改,省去了很多中间步骤,比如一开始省去了通过遗传办审批的30-60天的时间,后来又补办的材料——这是君实的VV116能在一个月内把实验做完的核心原因之一。
而在疫情的压力之下,研究机构、监管、伦理等等各方角色也都一路开绿灯,在推着国产新冠口服药前进。
但奥密克戎传染性强致病性弱的条件下,国内新冠口服药审批指导原则例强调的“重症改善率”,仍是阻碍很多新冠口服药的临床进度的一大难题。
03、监管的两难抉择
2022年2月17日,CDE发布的《新冠药物的临床指导原则》,在“有效性终点”的设计上,依旧是按照包括“住院或死亡患者比例”“恢复时间”等症状改善的终点。这也意味着,如果不按照“症状改善”的指标做临床终点,基本不会被获批做三期临床。
但EMI发表的VV116的研究结果里面提到了很现实的一点:用药组和常规干预组的患者全都没有发展成重症,这里面也就没有了研究的空间。因为奥密克戎的感染使得感染人群大多是无症状感染者和轻症,重症和住院人数占比极少。
这也是困扰很多新冠口服药研究者的难题。
辉瑞的新冠口服药Paxlovid当时针对的是德尔塔毒株,在美国特定人群中导致的住院率和重症率都较高,所以当时P药也有可以研究的空间,而FDA对新冠药物的审批,也很严格地以“改善重症、住院率”为主要终点。
一度有专家呼吁将“降低病毒载量”这一指标作为主要的临床终点,但这一指标又过于简单,防控专家吴尊友在前不久一次讲话中明确否决了这一做法:“病毒载量不属于三期临床试验的主要指标,三期临床试验指标的选择应当还是取决于对疾病严重情况的观察。病例在国内外都可以获得,需要与医院、医生进行联系合作。临床难做不应该是药监局降低审评标准的理由。”
此次EMI发表的研究结果,也仅仅是围绕“核酸转阴天数”去做的讨论,考虑到核酸阴性结果是通过病毒载量确定的,这次结果仍没有逃离病毒载量这一指标。
但是在和Paxlovid头对头实验中,采用了临床改善作为该临床试验的主要终点,但如果已经上市的P药在它的标准风险的三期临床试验EIPC-SR中在其临床主要终点"临床改善时间"这点上没有做出和安慰剂有差异的阳性结果,VV116只要做到非劣效结果获批,逻辑上很难自圆其说。
有人觉得跟一个在临床症状改善上没有做出阳性数据的药做临床改善的非劣对比,而不是采用P药获批的重症和死亡率这一主要终点指标,这个监管标准有些过低,但这也回到了一个问题:监管当时批准P药是基于临床改善吗?
这大概也是君实和各研究方在今年4月启动头对头临床研究的初衷之一。
国内其他新冠口服药研究者同样面对该问题,但并不是所有申办方都有如此条件去和辉瑞做头对头试验,比如P药的采购成本都不是一个小数目。
但也有另辟蹊径的。
先声药业在前日披露了一项关于暴露后预防的临床试验获批的通知,直接跳过了治疗效果,研究新冠口服药的预防效果。此前辉瑞的P药关于新冠预防的临床并没有成功,先声这个临床,有一些“迎难而上“的感觉,但同样也是当前新冠口服药审批环境下的一个选择。
在奥密克戎毒株大流行的情况下,降低病毒载量、降低传染率同样有着很重要意义。但药监作为药品质量守护的最后一道关卡,如何在“降低病毒载量”和“减轻症状”之间平衡,根据最新毒株奥密克戎的特点,也是监管部门的一个挑战。
本网页由机器采集生成,若侵权请及时联系删除。
原文链接:https://news.pedaily.cn/202205/492855.shtml